
This DNA mismatch repair protein plays an essential role in cancer research and diagnostic workflows. Scientists investigating Lynch syndrome and microsatellite instability depend on accurate detection methods. Understanding optimal conditions for sample preparation, dilution ranges, and incubation parameters determines experimental success.
Technical specifications indicate ideal dilution ranges between 1:2000 and 1:10000 for most cell lysates. Room temperature incubation for 1.5 hours produces consistent results. We examine antibody optimization techniques that address blocking strategies, detection methods, and troubleshooting approaches. Our structured methodology enables you to implement validated protocols while understanding the principles behind successful protein analysis in your laboratory workflows.
Key Takeaways
- Optimal dilution ranges for Western blotting fall between 1:2000 and 1:10000 depending on cell type and expression levels
- Room temperature incubation for 1.5 hours provides consistent detection results across multiple cell lines
- Proper sample preparation and blocking strategies significantly impact signal clarity and background reduction
- Understanding protein function helps interpret results in cancer research and Lynch syndrome diagnostics
- Validated protocols ensure reproducible detection across HeLa, HEK-293T, MCF-7, and other common cell lysates
- Technical specifications including buffer composition and preservatives affect antibody stability and performance
Introduction to PMS2 Rabbit Monoclonal Antibody
The PMS2 rabbit monoclonal antibody serves as a critical reagent for identifying DNA mismatch repair pathway disruptions in hereditary cancer syndromes. This specialized antibody enables researchers and clinical laboratories to detect PMS2 protein expression levels in tissue samples through Western blotting and immunohistochemical techniques. We recognize that understanding the molecular characteristics of PMS2 protein provides essential context for implementing optimal detection protocols.
As a fundamental component in cancer diagnostics, PMS2 antibody detection has revolutionized how we approach Lynch syndrome testing and related genetic screening programs. The specificity of rabbit monoclonal antibodies offers superior performance compared to traditional polyclonal alternatives. This enhanced precision makes PMS2 detection particularly valuable for laboratories conducting high-volume diagnostic testing.
What is PMS2?
PMS2 represents a 110 kDa mismatch repair endonuclease belonging to the MutL homolog family of proteins. The gene encodes 862 amino acids organized into distinct functional domains that work together to maintain genomic stability. This protein functions as an essential component in cellular DNA repair mechanisms that prevent mutation accumulation during cell division.
The molecular structure of PMS2 includes an N-terminal ATPase domain from the GHKL superfamily. Additionally, the protein contains a C-terminal endonuclease domain responsible for DNA cleavage activity. These structural elements enable PMS2 to recognize and process DNA replication errors effectively.
PMS2 forms the heterodimeric MutLα complex through interaction with MLH1 protein. This complex contains latent endonuclease activity that becomes stimulated through interactions with PCNA, RFC, MutSα, and ATP. The coordinated function of these molecular components creates a robust system for detecting and repairing DNA mismatches.
In normal cellular function, the mismatch repair pathway prevents approximately 1,000-fold increase in mutation rates. When PMS2 protein expression becomes lost or reduced, cells accumulate replication errors at microsatellite sequences throughout the genome. This microsatellite instability serves as a molecular signature for mismatch repair deficiency.
Significance of PMS2 in Cancer Research
Loss of PMS2 function through germline mutations constitutes one of four primary genetic defects causing Lynch syndrome. This autosomal dominant condition represents the most common form of hereditary colorectal cancer, affecting approximately 1 in 300 individuals. Patients with defective PMS2 expression demonstrate substantially elevated lifetime cancer risks across multiple organ systems.
The clinical significance of PMS2 extends beyond genetic screening into therapeutic decision-making. Tumors exhibiting microsatellite instability due to mismatch repair deficiency respond differently to certain chemotherapy agents and immunotherapy treatments. This makes PMS2 status determination a valuable colorectal cancer biomarker for personalized medicine approaches.
Mayo Clinic studies examining 535 microsatellite instability-high cases revealed important patterns in mismatch repair protein loss. Research showed that 90% of cases demonstrated loss of MLH1, MSH2, and/or MSH6 expression. Among the remaining cases, 70% exhibited isolated PMS2 loss, highlighting the protein’s unique diagnostic importance.
PMS2 antibody assists in microsatellite instability diagnosis alongside other mismatch repair markers. The coordinated analysis of all four mismatch repair proteins provides comprehensive screening for hereditary cancer syndromes. This multi-marker approach has become standard practice in pathology laboratories conducting Lynch syndrome testing.
| PMS2 Characteristic | Clinical Relevance | Detection Application |
|---|---|---|
| 110 kDa protein size | Distinct Western blot band identification | Molecular weight confirmation in protein analysis |
| Forms MutLα complex with MLH1 | Co-expression pattern analysis required | Coordinated testing with MLH1 antibody |
| Isolated loss in 70% of remaining MSI cases | Unique diagnostic marker for specific Lynch syndrome subset | Essential for complete hereditary cancer screening panels |
| Nuclear localization in normal cells | Expected staining pattern for functional protein | Quality control reference for immunohistochemistry |
Patients carrying PMS2 mutations face increased lifetime risks for multiple malignancies. Colorectal cancer risk reaches 20-25%, while endometrial cancer risk approaches 15% in affected individuals. Additional cancer susceptibilities include gastric, ovarian, and urinary tract malignancies, necessitating comprehensive surveillance strategies.
The molecular signatures created by PMS2 deficiency enable immunohistochemical detection in tissue samples. This makes PMS2 rabbit monoclonal antibody an indispensable tool for screening programs in clinical pathology laboratories. We emphasize that accurate PMS2 detection forms a cornerstone of modern hereditary cancer syndrome diagnosis and patient management strategies.
Understanding Western Blotting Techniques
As one of the most reliable molecular pathology tools available, Western blotting delivers unparalleled specificity for protein detection in complex biological samples. We utilize this immunoblotting methodology to identify, quantify, and characterize proteins like PMS2 with exceptional precision. The technique combines protein analysis techniques with immunohistochemistry detection principles, creating a powerful platform for cancer research applications.
This method provides researchers with critical advantages when working with PMS2 antibodies. You gain high specificity through targeted antibody recognition, quantitative capacity for measuring expression levels, and molecular weight confirmation that ensures accurate protein identification. These benefits make Western blotting essential for laboratories conducting mismatch repair protein studies.
Fundamentals of the Immunoblotting Methodology
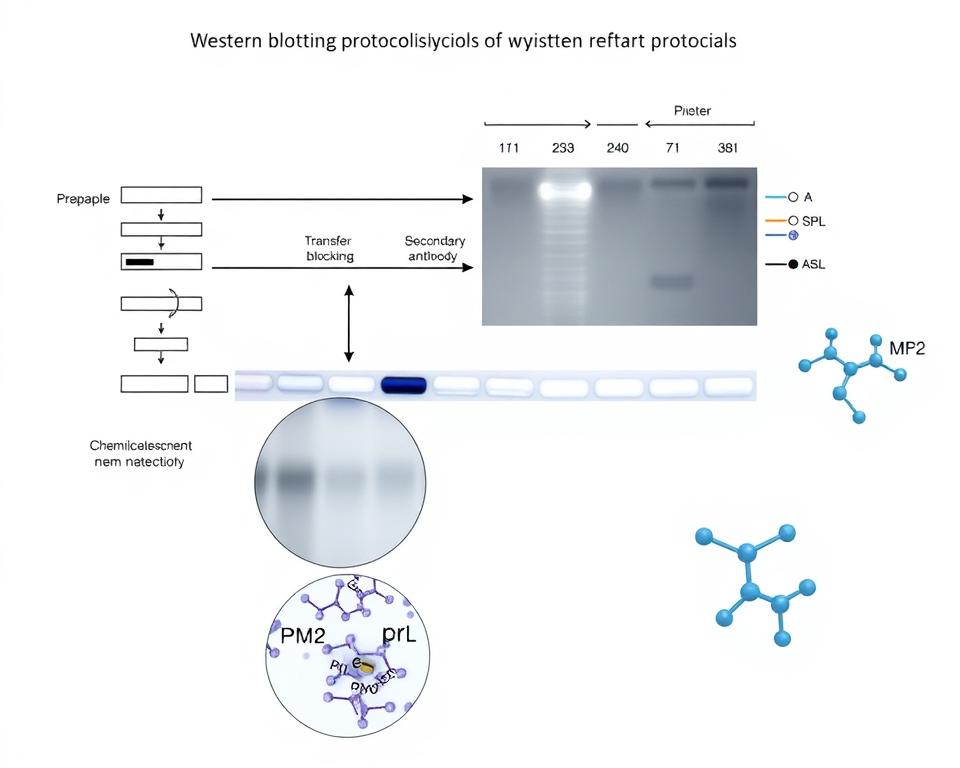
Western blotting represents a multi-stage analytical technique that separates proteins by size and detects specific targets through antibody binding. The process begins with protein separation using electrophoresis methods, specifically sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). This separation creates distinct protein bands based on molecular weight, with PMS2’s 110 kDa size providing clear differentiation from most cellular proteins.
Following separation, we transfer proteins from the gel onto a membrane matrix. Nitrocellulose and polyvinylidene difluoride (PVDF) membranes serve as the two primary options for this transfer step. The membrane provides an accessible surface where antibodies can bind to immobilized proteins, enabling subsequent detection steps.
The detection phase employs a two-antibody system that amplifies signal intensity. Primary antibodies, such as PMS2 rabbit monoclonal antibodies, recognize and bind to the target protein with exceptional specificity. Secondary antibodies conjugated to detection enzymes then bind to the primary antibody, creating a signal amplification cascade. For PMS2 detection, we typically use secondary antibodies conjugated to horseradish peroxidase (HRP), which catalyzes chemiluminescent reactions for visualization.
This technique stands apart from other molecular pathology tools through its combination of separation power and detection sensitivity. You can detect proteins present at nanogram quantities while simultaneously confirming their molecular weight. This dual capability proves invaluable when verifying PMS2 expression in clinical samples or research models.
Sequential Workflow for Optimal Protein Detection
The Western blotting process encompasses seven interconnected stages, each requiring careful optimization for successful PMS2 detection. Understanding these steps enables you to troubleshoot issues and achieve consistent results across experiments.
Stage 1: Sample Preparation and Protein Extraction
We begin by lysing cells or tissue samples using specialized buffers that preserve protein integrity. These buffers contain protease inhibitors that prevent protein degradation and phosphatase inhibitors that maintain post-translational modifications. Proper lysis ensures complete protein extraction while maintaining native protein structure.
Protein quantification follows extraction using Bradford or bicinchoninic acid (BCA) assays. These colorimetric methods determine total protein concentration, allowing you to load equal amounts across gel lanes. Equal loading prevents misinterpretation of expression differences and ensures accurate comparative analysis.
Stage 2: Gel Electrophoresis and Protein Separation
SDS-PAGE electrophoresis separates proteins based on molecular weight through a polyacrylamide gel matrix. The percentage of polyacrylamide determines separation resolution, with 8-12% gels optimal for proteins in PMS2’s size range. We apply electric current to drive negatively charged protein-SDS complexes through the gel, creating size-based separation.
Electrophoresis methods for Western blotting typically require 60-120 minutes depending on gel composition and voltage settings. Running gels at lower voltages produces sharper bands with better resolution, while higher voltages reduce run time but may compromise band quality.
Stage 3: Protein Transfer to Membrane
Electrotransfer moves separated proteins from the gel onto a membrane surface. We position the gel against the membrane in a transfer apparatus and apply perpendicular electric current. This current drives proteins out of the gel matrix and onto the membrane, where they remain immobilized for antibody detection.
| Transfer Parameter | Nitrocellulose Membrane | PVDF Membrane | Optimal for PMS2 |
|---|---|---|---|
| Transfer Time | 60-90 minutes | 90-120 minutes | 90 minutes |
| Voltage Setting | 100V constant | 100V constant | 100V constant |
| Protein Binding Capacity | 80-100 μg/cm² | 150-200 μg/cm² | PVDF preferred |
| Background Signal | Lower | Slightly higher | Both acceptable |
Stage 4: Membrane Blocking
Blocking prevents non-specific antibody binding by saturating the membrane with inert proteins. We typically use 5% non-fat dry milk or bovine serum albumin (BSA) in Tris-buffered saline with Tween-20 (TBST). This step reduces background signal and improves signal-to-noise ratios in final images.
Stage 5: Primary Antibody Incubation
Primary antibody application represents the critical specificity-determining step. For PMS2 detection, various lysates are subjected to antibody incubation at dilutions ranging from 1:2000 to 1:10000. We incubate membranes with diluted PMS2 antibody at room temperature for 1.5 hours or overnight at 4°C for enhanced sensitivity.
The antibody recognizes specific epitopes on the PMS2 protein, binding with high affinity while avoiding cross-reactivity with other cellular proteins. This specificity distinguishes Western blotting from less selective protein analysis techniques.
Stage 6: Secondary Antibody Application
Following primary antibody incubation and washing steps, we apply HRP-conjugated secondary antibodies. These antibodies recognize the primary antibody’s constant region, creating a detection complex. Typical secondary antibody incubations last 60 minutes at room temperature with dilutions between 1:5000 and 1:10000.
Stage 7: Signal Detection and Imaging
Detection utilizes enhanced chemiluminescence (ECL) substrates that react with HRP to produce light emission. We capture this light using charge-coupled device (CCD) cameras or X-ray film in imaging systems. The intensity of chemiluminescent signal correlates directly with protein abundance, enabling both qualitative identification and quantitative analysis.
Modern imaging systems provide digital output with linear detection ranges spanning several orders of magnitude. This dynamic range allows you to detect both highly abundant and low-expression proteins within the same experiment. For immunohistochemistry detection applications, this sensitivity proves essential when analyzing clinical specimens with variable PMS2 expression levels.
Understanding these fundamental steps establishes the foundation for implementing optimal PMS2-specific protocols. Each stage offers opportunities for optimization based on your specific experimental requirements and sample characteristics.
Importance of Using Antibodies in Western Blotting
In Western blotting applications, antibodies determine the critical parameters of assay performance, including sensitivity, specificity, and experimental reproducibility. These immunological reagents function as molecular recognition elements that bind target proteins with high precision. The choice of antibody type directly impacts the quality of data you obtain from your experiments.
We rely on antibodies to distinguish between closely related proteins in complex biological samples. Their binding characteristics establish whether your Western blot will produce clear, interpretable results or ambiguous data requiring troubleshooting. Understanding antibody classifications helps researchers select optimal tools for PMS2 detection protocols.
Molecular Recognition Through Monoclonal Antibodies
Monoclonal antibodies originate from single B-cell clones, producing identical immunoglobulin molecules that recognize one specific epitope on the target antigen. This clonal origin provides exceptional monoclonal antibody specificity that eliminates cross-reactivity concerns common with polyclonal preparations. Each molecule in a monoclonal preparation binds the same antigenic site with identical affinity.
The consistency of monoclonal antibodies enables protocol standardization across laboratories and experimental timepoints. You can trust that results obtained today will match those from experiments conducted months later. This reproducibility proves essential for longitudinal studies and multi-center research collaborations.
For PMS2 detection, monoclonal antibodies target specific domains within the protein structure. Common PMS2 antibody clone variants recognize epitopes in the N-terminal ATPase region or C-terminal endonuclease domain. These targeted binding sites ensure precise molecular identification without interference from structurally similar proteins.
Modern antibody production methods employ hybridoma technology or recombinant expression systems to generate monoclonal antibodies. These approaches guarantee batch-to-batch consistency that proves impossible with polyclonal antisera. The EP51 clone represents one well-characterized PMS2 antibody clone demonstrating high specificity in Western blotting applications.
Enhanced Performance of Rabbit-Derived Monoclonals
Rabbit monoclonal antibodies combine the specificity advantages of monoclonal technology with superior immune response characteristics. Rabbits generate higher affinity antibodies compared to traditional mouse hybridoma systems due to their more diverse antibody repertoire. This diversity enables recognition of smaller epitopes that mouse immune systems may overlook.
The enhanced affinity translates directly to improved sensitivity in Western blotting experiments. Rabbit monoclonals detect lower abundance proteins with greater signal-to-noise ratios than mouse counterparts. You can identify PMS2 in samples where expression levels remain below the detection threshold of other antibody types.
Recombinant technology employed in rabbit monoclonal production enables epitope-specific antibody generation. We can target conserved protein regions that maintain structure across species, ensuring broad reactivity. This characteristic proves valuable when comparing PMS2 expression patterns across different model organisms or tissue types.
| Antibody Characteristic | Mouse Monoclonal | Rabbit Monoclonal | Polyclonal |
|---|---|---|---|
| Epitope Recognition | Single epitope | Single epitope | Multiple epitopes |
| Affinity Level | Moderate to high | High to very high | Variable |
| Batch Consistency | Excellent | Excellent | Poor |
| Sensitivity in Western Blot | Good | Superior | Moderate |
| Species Reactivity | Limited | Broad | Broad but variable |
The IgG isotype characteristic of rabbit monoclonals provides stable binding properties across various experimental conditions. These immunological reagents maintain functionality through pH variations, temperature fluctuations, and different buffer compositions. Such stability ensures reliable performance regardless of minor protocol adjustments.
Production consistency represents another critical advantage of modern rabbit antibody production methods. Recombinant approaches eliminate animal-to-animal variation inherent in traditional immunization protocols. Each production batch maintains identical binding characteristics, ensuring your results remain comparable across the entire duration of your research project.
When you select rabbit monoclonal antibodies for PMS2 detection, you invest in tools engineered for optimal Western blotting performance. The combination of high specificity, superior sensitivity, and exceptional reproducibility supports confident data interpretation and publication-quality results.
Selection Criteria for PMS2 Rabbit Monoclonal Antibody
Researchers face multiple considerations when identifying optimal PMS2 rabbit monoclonal antibodies for their specific diagnostic applications. The selection process requires systematic evaluation of technical parameters that determine experimental reliability and reproducibility. We guide you through essential criteria that separate high-performance reagents from suboptimal alternatives.
Understanding these selection factors ensures you invest in antibodies that deliver consistent results across your Western blotting protocols. The right choice minimizes troubleshooting time and maximizes data quality in your cancer research applications.
Factors to Consider When Choosing Antibodies
Several critical parameters demand careful assessment before selecting your PMS2 Rabbit Monoclonal Antibody for Western blotting applications. We recommend evaluating each factor systematically to match reagent capabilities with your experimental requirements.
Antibody validation stands as the primary selection criterion for any cancer diagnostics reagent. You should examine validation data demonstrating performance specifically in Western blotting applications. Antibodies validated only for immunohistochemistry may not transfer successfully to immunoblot protocols.
Confirm that validation includes appropriate positive controls showing clear PMS2 detection at the expected 110 kDa molecular weight. Validation should demonstrate consistent band detection across multiple cell lines and tissue types.
- Epitope specificity: Review immunogen information to understand which PMS2 region the antibody recognizes. Antibodies raised against synthetic peptides corresponding to unique PMS2 sequences minimize cross-reactivity with related MutL family proteins including MLH1, MLH3, and PMS1.
- Species reactivity: Confirm human protein recognition for clinical sample applications. This becomes particularly important for translational research involving patient-derived specimens.
- Dilution range recommendations: Assess suggested working concentrations that balance sensitivity with background signal levels. Optimal dilution ranges indicate thorough manufacturer testing.
- Storage stability: Examine formulation composition and storage requirements ensuring the antibody remains functional throughout your experimental timeline.
- Clone identification: Document clone numbers for reproducibility and literature comparison purposes.
Nuclear visualization in positive control tissues such as tonsil, appendix, and colon sections confirms proper antibody function. These tissues express high PMS2 levels and serve as reliable validation controls.
Specificity confirmation through knockout or knockdown studies provides additional confidence in antibody validation quality. These experiments demonstrate that signal disappears when PMS2 expression is eliminated.
Differences Between Monoclonal and Polyclonal Antibodies
Understanding the fundamental distinctions between monoclonal and polyclonal antibodies informs better selection decisions for your specific application. Each antibody type offers unique advantages and limitations that impact Western blotting performance.
Monoclonal antibodies recognize single epitopes on the target protein. This specificity provides exceptional discrimination between structurally similar proteins. For PMS2 Western blotting, monoclonal antibodies typically deliver superior performance due to high specificity essential for distinguishing PMS2 from MLH1 with which it forms functional complexes.
Batch-to-batch consistency represents another critical advantage of monoclonal antibodies. Once you establish optimal protocols, results remain reproducible across antibody lots. This consistency proves invaluable for long-term studies and quantitative applications.
However, single epitope recognition means protein modifications or conformational changes near the binding site may abolish detection. This limitation requires consideration when studying post-translational modifications.
Polyclonal antibodies recognize multiple epitopes across the target protein. This multi-epitope binding offers robustness against epitope alterations and potentially higher overall signal due to multiple binding events per target molecule.
| Characteristic | Monoclonal Antibodies | Polyclonal Antibodies |
|---|---|---|
| Epitope Recognition | Single specific epitope | Multiple epitopes across protein |
| Specificity Level | Very high – minimal cross-reactivity | Moderate – potential cross-reactivity |
| Batch Consistency | Excellent reproducibility | Variable between production lots |
| Signal Sensitivity | Consistent signal strength | Potentially stronger due to multi-binding |
| Modification Tolerance | Sensitive to epitope changes | Robust against single epitope alterations |
The structural similarity between PMS2 and other MutL family proteins makes epitope specificity particularly important. Monoclonal antibodies raised against unique PMS2 sequences provide the discrimination necessary for accurate protein quantification.
We recommend monoclonal antibodies for most cancer diagnostics reagent applications involving PMS2 Western blotting. The combination of high specificity, batch consistency, and reliable performance justifies their selection for both research and diagnostic protocols.
Preparing Samples for Western Blotting
The quality of your Western blot results depends heavily on how you prepare your protein samples before analysis. We recognize that sample quality control establishes the foundation for detecting PMS2 protein with accuracy and reproducibility. Every step from initial extraction to final loading impacts your experimental outcome.
Proper sample handling prevents protein degradation and preserves the native state of PMS2. This attention to detail becomes especially critical when working with nuclear proteins like PMS2, which require specific extraction protocols to achieve optimal detection sensitivity.
Critical Foundation for Reliable Detection
Sample preparation represents the most influential factor in Western blotting success. Poor preparation techniques introduce variability that no amount of downstream optimization can correct. We emphasize that investing time in proper sample treatment yields consistent, interpretable results.
Your choice of cell lines or tissue specimens directly affects PMS2 signal strength. Positive control samples should demonstrate robust PMS2 expression to validate your detection system. Cell lines showing reliable PMS2 expression include:
- HeLa cells (cervical carcinoma)
- HEK-293T cells (embryonic kidney)
- PC-3 cells (prostate adenocarcinoma)
- MCF-7 cells (breast adenocarcinoma)
- A431 cells (epidermoid carcinoma)
- Caco-2 cells (colorectal adenocarcinoma)
Cell lysis methodology significantly influences protein recovery rates. Standard protein extraction methods employ buffer systems designed to solubilize cellular proteins while maintaining structural integrity. We recommend radioimmunoprecipitation assay (RIPA) buffer for comprehensive protein extraction.
The optimal RIPA buffer composition includes 50 mM Tris-HCl at pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS. Supplement this buffer with complete protease inhibitor cocktails to prevent enzymatic degradation during extraction. Add phosphatase inhibitors when examining post-translational modifications.
Maintain samples on ice throughout the extraction process. This temperature control minimizes protease activity and preserves protein integrity. Use mechanical disruption techniques such as repeated pipetting or brief vortexing to ensure complete cell lysis.
Optimizing Sample Treatment Protocols
After lysis, centrifuge samples at 12,000-15,000 x g for 10-15 minutes at 4°C. Collect the supernatant containing soluble proteins while discarding the pellet of cellular debris. This clarification step removes insoluble material that could interfere with electrophoresis.
PMS2 localizes primarily to the nucleus, making nuclear-cytoplasmic fractionation a valuable technique for enhanced detection. This approach enriches nuclear proteins while removing abundant cytoplasmic proteins. The result is cleaner blots with stronger PMS2 signals and reduced background interference.
Protein quantification ensures equal loading across gel lanes, a requirement for accurate comparative analysis. Molecular pathology tools such as Bradford or bicinchoninic acid (BCA) assays provide reliable measurements. We typically load 20-40 μg of total protein per lane for optimal PMS2 detection.
Prepare samples in Laemmli buffer containing reducing agents like β-mercaptoethanol or dithiothreitol. Heat samples at 95-100°C for 5 minutes to denature proteins completely. This treatment ensures linear migration during electrophoresis and facilitates antibody recognition of PMS2 epitopes.
Storage practices profoundly impact protein stability over time. Aliquot your samples into single-use volumes immediately after preparation. This practice eliminates repeated freeze-thaw cycles that progressively degrade protein integrity and compromise experimental reproducibility.
Store aliquoted samples at -80°C for long-term preservation. Never refreeze samples after thawing, as each freeze-thaw cycle reduces protein quality. When planning experiments requiring pathology laboratory supplies, ensure adequate storage capacity for properly preserved samples.
Quality control checkpoints throughout sample preparation verify that your proteins remain intact and suitable for analysis. Visual inspection of lysates should reveal clear, non-viscous solutions. Excessive viscosity indicates incomplete lysis or genomic DNA contamination requiring additional treatment.
Document your sample quality control measurements including protein concentrations and sample volumes. This record-keeping enables troubleshooting if unexpected results appear during detection. Consistent documentation also facilitates protocol optimization and laboratory compliance requirements.
Optimizing Antibody Dilution Ratios
Antibody dilution optimization serves as a critical balance point between signal intensity, background noise, and cost-effectiveness in PMS2 detection protocols. Finding the right concentration ensures you obtain clear, reproducible results while maximizing reagent efficiency. We recommend approaching dilution optimization systematically to establish robust protocols tailored to your specific experimental conditions.
The process of protocol optimization requires understanding both theoretical principles and practical variables that influence antibody performance. Different laboratory setups, equipment variations, and sample types necessitate customized approaches rather than universal application of published protocols.

Starting Points for PMS2 Antibody Dilution
Western blot dilution ranges for PMS2 rabbit monoclonal antibodies typically span 1:2000 to 1:10000, with 1:5000 representing a commonly used starting dilution. These ranges provide flexibility for different experimental requirements and antibody formats.
Ready-to-use formulations require no dilution adjustment and offer convenience for standardized applications. Concentrated stock solutions, however, demand empirical testing to determine optimal working concentrations.
For concentrated PMS2 antibody clone preparations, begin with manufacturer recommendations as your baseline. We suggest conducting dilution series experiments to identify the ideal concentration for your specific system.
Systematic Approach to Dilution Testing
Perform dilution series experiments testing multiple concentrations simultaneously using identical sample sets. This parallel approach saves time and ensures direct comparability between conditions.
Test the following dilution series as starting points:
- 1:1000 – Higher antibody concentration for low-abundance targets
- 1:2000 – Moderate concentration balancing signal and background
- 1:5000 – Standard starting dilution for most applications
- 1:10000 – Higher dilution for abundant targets or high-affinity antibodies
Evaluate each dilution for signal intensity at the 110 kDa PMS2 band, background signal across the membrane, and overall signal-to-noise ratio. The optimal dilution produces strong specific signal with minimal non-specific binding.
For most high-affinity rabbit monoclonal antibodies, the optimal range typically falls between 1:2000 and 1:5000. Some protocols recommend 1:50 dilution for highly concentrated antibody preparations, though this requires careful validation to prevent background issues.
Critical Variables Affecting Dilution Selection
Multiple factors influence optimal antibody concentration beyond the antibody itself. Understanding these variables enables informed decision-making during protocol optimization.
Monoclonal antibody specificity directly correlates with dilution capacity. Higher affinity antibodies maintain specific binding at greater dilutions, enabling reagent conservation without sensitivity loss. This characteristic makes rabbit monoclonal antibodies particularly economical for routine applications.
Target protein abundance significantly affects required antibody concentration. PMS2 expression levels vary across cell types, with mismatch repair-proficient cells expressing moderate levels while deficient cells show absent or dramatically reduced expression.
| Factor | Impact on Dilution | Optimization Strategy |
|---|---|---|
| Antibody Affinity | Higher affinity enables greater dilution | Test extended dilution range for high-affinity clones |
| Target Abundance | Low expression requires higher concentration | Adjust based on cell line characteristics |
| Detection Sensitivity | Enhanced systems allow higher dilution | Match antibody concentration to substrate type |
| Membrane Type | PVDF may require higher concentration | Optimize separately for each membrane format |
Detection system sensitivity influences dilution optimization substantially. Enhanced chemiluminescence substrates enable higher primary antibody dilutions compared to standard ECL reagents due to improved signal amplification capabilities.
Membrane type affects antibody binding kinetics and optimal concentrations. PVDF membranes generally require slightly higher antibody concentrations than nitrocellulose due to higher protein binding capacity and different surface properties.
Incubation Conditions and Timing
Incubation parameters work synergistically with antibody concentration to determine final results. Standard protocols recommend incubation at room temperature for 1.5 hours or at 4°C overnight for primary antibody binding.
Room temperature incubation provides faster results and works well for optimized protocols. Overnight incubation at 4°C can enhance signal intensity for challenging targets or when using higher dilutions.
We recommend optimizing dilution conditions specific to your experimental system rather than universally applying literature protocols. Subtle differences in reagents, equipment, and samples affect optimal parameters significantly.
Practical Implementation Guidelines
Document your optimization experiments thoroughly, recording all variables including lot numbers, incubation times, and blocking conditions. This documentation enables troubleshooting and protocol refinement over time.
Consider reagent economy when selecting final working dilutions. If multiple dilutions produce acceptable results, choose the higher dilution to maximize antibody lifespan and reduce costs without compromising data quality.
Validate your optimized protocol across multiple experimental runs before adopting it as your standard procedure. Consistency across replicates confirms robustness and reliability for routine applications.
Blocking and Incubation Steps
Effective blocking and careful incubation timing establish the foundation for accurate DNA mismatch repair protein detection in Western blotting applications. These procedural phases directly determine the specificity of your results and the quality of signal you obtain during immunohistochemistry detection. We provide comprehensive protocols that balance efficiency with reliability.
The relationship between blocking effectiveness and antibody performance cannot be overstated. Proper execution of these steps separates successful protein detection from problematic background interference.
The Critical Role of Blocking in Protein Detection
Blocking serves as the primary defense against unwanted background signals in Western blot analysis. The membrane surface contains numerous protein binding sites that remain available after electrophoretic transfer completes. Without adequate blocking, antibodies attach to these sites rather than exclusively binding target proteins.
We recommend saturating the membrane with blocking buffer for 60 minutes at room temperature. This duration allows complete coverage of available binding sites, establishing effective non-specific binding prevention throughout the membrane surface.
The blocking buffer composition significantly influences DNA mismatch repair protein detection outcomes. Standard protocols utilize 5% non-fat dry milk dissolved in Tris-buffered saline with 0.1% Tween-20 (TBST). This economical solution provides excellent blocking capacity for most applications.
Consider these blocking buffer alternatives for specialized applications:
- Bovine serum albumin (BSA): Substitute 5% BSA when detecting phosphorylated proteins, as milk proteins contain kinases that interfere with phospho-epitope recognition
- Commercial blocking solutions: Pre-formulated buffers offer consistency across experiments but increase procedural costs
- Serum-based blockers: Normal serum from secondary antibody host species reduces cross-reactivity in complex detection systems
For PMS2 detection, milk-based blocking typically delivers optimal results. The protein rarely requires phosphorylation-specific analysis, making standard milk buffers the preferred choice.
Maintain gentle agitation throughout blocking using orbital shakers set to 30-50 rpm. This movement ensures uniform buffer distribution across the membrane surface, preventing localized blocking deficiencies that create spotty backgrounds.
Optimizing Antibody Incubation Parameters
Primary antibody incubation represents the moment when target protein detection occurs. The conditions you establish during this phase directly control binding efficiency and specificity. We examine multiple parameters that collectively determine detection success.
Dilute your PMS2 rabbit monoclonal antibody in blocking buffer rather than plain TBST. This approach maintains blocking capacity during incubation, continuously preventing non-specific binding as antibody molecules search for target epitopes.
Temperature and duration function as interconnected variables in antibody binding conditions. Room temperature incubation for 1.5 hours provides the optimal balance for PMS2 detection. This timing allows high-affinity monoclonal antibodies sufficient opportunity to locate and bind target proteins without extending protocols unnecessarily.
| Incubation Parameter | Standard Condition | Alternative Option | Application Context |
|---|---|---|---|
| Temperature | Room temperature (20-25°C) | 4°C overnight | Low-abundance targets or lower-affinity antibodies |
| Duration | 1.5 hours | 8-16 hours | Enhanced sensitivity requirements |
| Agitation Speed | 30-50 rpm | Gentle rocking | Fragile membranes or large antibody complexes |
| Antibody Volume | Minimum coverage (5-10 mL) | Excess volume (20-30 mL) | Antibody conservation versus uniform distribution |
The choice between room temperature and cold incubation depends on your specific experimental requirements. Room temperature protocols offer convenience and typically sufficient binding for DNA mismatch repair protein detection. Cold overnight incubation may enhance signal when working with particularly low protein concentrations.
Continuous gentle agitation remains essential throughout primary antibody incubation. Stagnant conditions create concentration gradients where antibody depletion occurs in membrane contact zones. Movement replenishes antibody molecules across the surface, ensuring uniform antibody binding conditions throughout the detection area.
Following primary antibody exposure, perform thorough washing to remove unbound molecules. We recommend five washes with TBST, each lasting 5-10 minutes with gentle agitation. Inadequate washing allows residual primary antibody to remain on the membrane surface, where it binds secondary antibody non-specifically and elevates background signals.
The washing buffer should contain 0.1% Tween-20 to disrupt weak hydrophobic interactions while preserving specific antibody-antigen binding. Higher detergent concentrations risk removing specifically bound antibodies, particularly when dealing with lower-affinity interactions.
These blocking and incubation protocols establish reliable conditions for immunohistochemistry detection across various experimental contexts. Consistency in executing these steps produces reproducible results that accurately reflect DNA mismatch repair protein expression levels in your samples.
Detection Methods for Western Blotting
The visualization step in Western blotting represents the final and most critical phase for PMS2 protein detection. Your choice of detection methodology directly influences sensitivity, dynamic range, and quantification accuracy. We examine the most effective approaches to ensure reliable results when working with this important colorectal cancer biomarker.
Detection systems transform antibody-antigen binding into measurable signals through various mechanisms. Each method offers distinct advantages depending on your experimental requirements and available equipment. Understanding these differences helps you optimize your protocol for PMS2 analysis.
Enzyme-Linked Detection Systems
Enzyme-conjugated secondary antibodies provide the foundation for most Western blot detection protocols. Horseradish peroxidase (HRP) conjugated to anti-rabbit IgG delivers exceptional performance for PMS2 detection due to its high catalytic efficiency. We recommend applying HRP-conjugated secondary antibodies at a 1:10,000 dilution in blocking buffer for one hour at room temperature.
This cancer diagnostics reagent catalyzes the oxidation of luminol in the presence of hydrogen peroxide. The reaction produces light emission that CCD cameras or film can detect. The process creates measurable signals proportional to the amount of target protein present on the membrane.
Chemiluminescence detection has become the gold standard for protein visualization in research laboratories. The method combines high sensitivity with broad accessibility. Most facilities already possess the necessary imaging equipment to capture chemiluminescent signals effectively.
Enhanced Chemiluminescence Substrate Selection
Standard ECL reagents generate adequate sensitivity for moderate to high abundance proteins. These formulations produce signals lasting one to two hours, providing sufficient time for image capture. However, the substrate you select significantly impacts your detection capabilities.
Enhanced ECL formulations incorporate signal amplification methods that extend detection duration and increase sensitivity 10 to 100-fold. These advanced substrates enable detection of low-abundance targets or highly diluted primary antibodies. For PMS2 detection in cell lysates with normal expression levels, standard ECL typically provides sufficient sensitivity.
When analyzing PMS2 expression in heterozygous mutation carriers, enhanced ECL substrates improve accuracy. These samples show reduced but not absent expression of this critical colorectal cancer biomarker. The enhanced sensitivity allows for better quantification of subtle differences in protein levels.
| Detection Method | Sensitivity Level | Dynamic Range | Signal Duration | Best Application |
|---|---|---|---|---|
| Standard ECL | Picogram range | 2-3 orders magnitude | 1-2 hours | Moderate to high abundance proteins |
| Enhanced ECL | Femtogram range | 3-4 orders magnitude | 4-24 hours | Low abundance targets, diluted antibodies |
| Fluorescent Detection | Low picogram range | 4-5 orders magnitude | Stable, no decay | Multiplexing, precise quantification |
| Colorimetric Detection | Nanogram range | 1-2 orders magnitude | Permanent | Simple documentation, teaching labs |
Alternative Detection Approaches
Fluorescent detection offers advantages for specific research applications requiring advanced capabilities. Fluorophore-conjugated secondary antibodies or infrared dyes provide wider linear dynamic ranges for quantification. This method also enables multiplexing capability for simultaneous detection of multiple targets on a single membrane.
The stability of fluorescent signals represents another key benefit. Unlike chemiluminescence detection, fluorescent signals do not decay over time. You can re-image membranes days or weeks after the initial detection without signal loss.
However, fluorescent detection requires specialized imaging equipment with appropriate excitation sources and emission filters. The initial investment costs exceed those for standard chemiluminescence detection systems. Additionally, fluorescent methods typically show lower sensitivity compared to enhanced ECL for single-target applications.
Selecting the Optimal Method for PMS2 Analysis
For routine PMS2 Western blotting, we recommend HRP-based chemiluminescence using enhanced ECL substrates. This combination provides an optimal balance of sensitivity, cost-effectiveness, and equipment accessibility. Most research laboratories already possess the necessary imaging systems for this approach.
When your research requires multiplexing PMS2 with other mismatch repair proteins, fluorescent detection becomes more appropriate. The ability to detect MLH1, MSH2, MSH6, and PMS2 simultaneously saves time and sample material. This approach proves particularly valuable when working with limited clinical specimens or this important cancer diagnostics reagent application.
Consider your experimental goals when selecting detection methods. If you need simple documentation of PMS2 presence or absence, standard ECL suffices. For quantitative analysis comparing expression levels across multiple samples, enhanced ECL or fluorescent detection provides superior accuracy. The signal amplification methods in enhanced substrates deliver the sensitivity needed for precise measurements.
Modern imaging systems with cooled CCD cameras capture chemiluminescent signals with exceptional sensitivity and broad dynamic range. These systems allow signal integration over extended exposure times, improving detection of weak signals. Software analysis tools facilitate quantification and normalization against loading controls.
Troubleshooting Common Issues in Western Blotting
Laboratory professionals frequently encounter specific challenges when working with PMS2 Rabbit Monoclonal Antibody in Western blot protocols. These technical difficulties can compromise experimental results and require systematic problem-solving approaches. Understanding common issues and their solutions ensures reliable data generation and supports effective protocol optimization.
We provide comprehensive troubleshooting guidance to address the most prevalent problems in PMS2 detection workflows. Implementing quality control measures throughout the experimental process helps identify issues early and prevents wasted time and resources. This systematic approach transforms technical challenges into opportunities for improved methodology and more consistent results.
Addressing Weak Signal Problems
Weak or absent signals at the expected 110 kDa position represent one of the most common frustrations in Western blotting. Multiple factors contribute to this issue, requiring sequential evaluation to identify the root cause. Systematic troubleshooting saves time and ensures you address the actual problem rather than treating symptoms.
First, assess protein loading by examining loading control bands such as β-actin, GAPDH, or PCNA. Weak loading controls indicate insufficient protein quantity in your samples. You should increase sample loading volume or concentrate your lysate before running the gel.
Second, evaluate your PMS2 Rabbit Monoclonal Antibody dilution ratio. Excessive dilution reduces binding site occupancy and diminishes signal intensity. Prepare fresh antibody solutions at higher concentrations and re-test your samples.
Third, examine incubation conditions carefully. Insufficient incubation time or improper temperature prevents adequate antibody-antigen binding. We recommend extending primary antibody incubation to overnight at 4°C for optimal results.
Detection reagent performance significantly impacts signal generation. Expired ECL substrate or inadequate mixing reduces chemiluminescent output. Always use fresh, properly prepared substrate and ensure thorough membrane coverage during application.
Antibody integrity affects detection capability directly. Improper storage or excessive freeze-thaw cycles damage antibody structure and reduce binding efficiency. Store your pathology laboratory supplies according to manufacturer specifications and avoid repeated thawing.
- Verify protein loading with loading controls
- Test multiple antibody concentrations systematically
- Extend incubation times for challenging samples
- Replace detection reagents if expiration dates have passed
- Monitor antibody storage conditions continuously
Include positive control samples with known PMS2 expression in every experiment. HeLa and MCF-7 cell lines provide reliable reference points for validating your troubleshooting efforts. These controls confirm whether issues stem from antibody performance or sample-specific factors.
Solutions for Non-Specific Binding
High background signal across the membrane or additional bands at incorrect molecular weights indicate non-specific binding issues. These problems compromise data interpretation and require targeted intervention strategies. Protocol optimization addresses non-specific binding through multiple complementary approaches.
Blocking conditions require careful optimization for PMS2 Rabbit Monoclonal Antibody applications. Insufficient blocking leaves binding sites available for non-specific antibody adhesion. Increase blocking time to one or two hours at room temperature.
You can also raise blocking agent concentration to five to ten percent. This enhanced blocking creates more effective barriers against non-specific interactions without interfering with target antigen detection.
Antibody concentration directly influences binding specificity. Excess antibody promotes non-specific adhesion to membrane surfaces and proteins. Test higher dilution ratios such as 1:10,000 or greater to reduce background signal.
| Problem Indicator | Root Cause | Recommended Solution | Expected Outcome |
|---|---|---|---|
| High background across membrane | Insufficient blocking | Extend blocking to 2 hours with 5-10% blocking agent | Clear background with distinct bands |
| Multiple non-specific bands | Excessive antibody concentration | Test dilutions from 1:5,000 to 1:15,000 | Single band at 110 kDa |
| Uniform membrane staining | Inadequate washing | Increase to 6 washes, 15 minutes each | Reduced overall background |
| Faint smearing patterns | Temperature-related binding | Switch to 4°C overnight incubation | Sharp, defined protein bands |
Washing stringency affects background levels significantly. Insufficient washing fails to remove unbound antibody molecules from membrane surfaces. We recommend increasing wash frequency to five or six cycles and extending duration to ten to fifteen minutes per wash.
Adding 0.2 to 0.5 percent Tween-20 to your wash buffer enhances detergent strength. This adjustment helps disrupt weak non-specific interactions while preserving specific antibody-antigen binding.
Incubation temperature influences binding specificity for certain antibody clones. Room temperature incubation may increase non-specific binding with PMS2 Rabbit Monoclonal Antibody. Switching to 4°C overnight incubation often resolves this issue completely.
Quality control measures must include appropriate positive and negative controls in every experiment. Positive controls validate that your pathology laboratory supplies and reagents function correctly. Negative controls confirm that observed bands represent specific PMS2 detection rather than experimental artifacts.
Document all troubleshooting steps and results systematically. This practice builds institutional knowledge and accelerates problem resolution in future experiments. Your documentation supports continuous improvement in laboratory protocols and maintains consistency across research teams.
Case Studies: Successful Applications of PMS2
Documented case studies reveal how PMS2 antibody protocols deliver reliable results in diagnostic laboratories. These real-world applications provide essential validation for clinical diagnostics applications and research methodologies. We examine representative examples that demonstrate effective PMS2 detection across diverse experimental contexts.
Clinical laboratories worldwide have integrated PMS2 antibody testing into comprehensive screening programs. The inclusion of PMS2 alongside MLH1, MSH2, and MSH6 creates a four-antibody panel approach for Lynch syndrome testing. This comprehensive strategy identifies mismatch repair deficiencies that single or dual-antibody approaches might miss.
Medical institutions report significant improvements in diagnostic accuracy when implementing expanded antibody panels. Studies show that adding PMS2 testing increases detection rates for hereditary cancer syndromes. The identification of isolated PMS2 loss cases represents a critical advancement in colorectal cancer biomarker screening protocols.
Examples in Cancer Research
Lynch syndrome testing protocols demonstrate the practical value of PMS2 antibody applications. Mayo Clinic research analyzing 535 cases with high microsatellite instability revealed important patterns. The data showed that 90% of tumors demonstrated loss of MLH1, MSH2, or MSH6 expression in various combinations.
The remaining cases provided particularly valuable insights into isolated protein loss. Approximately 70% of these remaining tumors showed isolated PMS2 loss as the only mismatch repair deficiency. This finding established PMS2 testing as essential rather than optional in comprehensive screening programs.
Clinical characteristics associated with PMS2 loss present distinct patterns. Patients with isolated PMS2-deficient tumors typically receive diagnosis at younger ages, with median ages ranging from 40 to 50 years. These tumors predominantly occur in right-sided colon locations, representing approximately 10-15% of all mismatch repair-deficient colorectal cancers.
Research applications extend beyond colorectal cancer into other malignancies. Endometrial carcinomas represent the most common non-colorectal cancers in hereditary cancer syndrome patients. Concurrent MLH1 and PMS2 loss constitutes the most frequently observed immunohistochemical abnormality in these cases, demonstrating PMS2 antibody relevance across multiple cancer types.
Insights from Scientific Studies
Western blot analysis provides mechanistic understanding of mismatch repair pathway function. Research examining PMS2 variants of uncertain significance utilized Western blotting to assess protein stability. Studies on the PMS2 p.Asn335Ser variant demonstrated maintained protein expression levels despite lacking ATPase activity, revealing unexpected functional relationships within DNA repair mechanisms.
These findings directly impact patient management through improved variant classification. Genetic counseling decisions rely on accurate determination of whether specific PMS2 variants represent pathogenic mutations. Western blot data contributes essential evidence for distinguishing benign polymorphisms from disease-causing alterations.
Translational research applications demonstrate PMS2 antibody utility in therapy selection. Tumors exhibiting mismatch repair deficiency show enhanced sensitivity to immune checkpoint inhibitors. This sensitivity results from high tumor mutational burden and increased neoantigen presentation, making PMS2 status a valuable predictive biomarker.
Quantitative Western blot analysis enables patient stratification for immunotherapy clinical trials. Research teams measure PMS2 expression levels to identify candidates most likely to benefit from specific treatment approaches. This application connects diagnostic testing with personalized medicine strategies.
The following table summarizes key findings from representative PMS2 application studies:
| Application Context | Key Finding | Clinical Significance | Detection Method |
|---|---|---|---|
| Comprehensive Screening Panels | 90% detection rate for MMR deficiency with four-antibody approach | Improved Lynch syndrome identification accuracy | Immunohistochemistry with PMS2, MLH1, MSH2, MSH6 |
| Isolated PMS2 Loss Cases | 10-15% of MMR-deficient tumors show only PMS2 absence | Identifies distinct molecular subgroup requiring specific genetic testing | Western blot and IHC correlation |
| Variant Characterization | p.Asn335Ser maintains expression without ATPase activity | Informs variant classification and genetic counseling decisions | Quantitative Western blot analysis |
| Immunotherapy Prediction | PMS2 deficiency correlates with checkpoint inhibitor response | Enables patient selection for targeted immunotherapy trials | Expression level quantification via Western blot |
CRISPR-Cas9 gene editing experiments benefit from PMS2 Western blot validation. Research teams targeting DNA repair pathways use PMS2 detection to confirm successful protein knockout or knockdown. These validation studies ensure experimental models accurately represent intended genetic modifications.
The versatility demonstrated across these applications underscores the importance of optimized protocols. Reliable, reproducible results depend on proper antibody selection, appropriate dilution ratios, and consistent experimental conditions. Each application context may require specific optimization adjustments while maintaining core protocol principles.
Comparing PMS2 with Other Antibodies
Comparing PMS2 antibodies with other mismatch repair protein antibodies reveals critical differences that guide optimal experimental design. We examine the functional relationships between these proteins to help you select the most appropriate antibody for your specific research or diagnostic application. Understanding these connections enables more accurate interpretation of Western blot results and improves diagnostic precision.
The mismatch repair pathway relies on four primary DNA mismatch repair proteins working in coordinated pairs. Each protein exhibits distinct characteristics that influence monoclonal antibody specificity and detection patterns. These relationships form the foundation for effective antibody panel selection in both research and clinical settings.
Similarities and Differences with Other Antibodies
PMS2 and MLH1 antibodies recognize proteins that form the MutLα heterodimeric complex, functioning together in the mismatch repair pathway. MLH1 serves as the dominant partner in this relationship, with PMS2 requiring MLH1 binding for protein stability. This functional dependency creates distinctive detection patterns that inform diagnostic strategies.
MLH1 deficiency causes concurrent PMS2 loss due to protein destabilization. In contrast, isolated PMS2 deficiency leaves MLH1 expression intact. This asymmetric relationship becomes evident in Western blot analysis when detecting both proteins simultaneously.
MLH1 germline mutations or MLH1 promoter hypermethylation both cause dual protein loss. However, isolated PMS2 loss specifically indicates PMS2 genetic alterations. These distinct patterns enable differentiation between various genetic scenarios and guide subsequent molecular testing.
MSH2 and MSH6 antibodies detect mismatch repair pathway proteins forming the alternative MutSα complex. This complex functions upstream of MutLα in mismatch recognition. MSH2 demonstrates similar dominant partner characteristics to MLH1 in its relationship with MSH6.
MSH2 deficiency causes MSH6 destabilization and concurrent loss. Isolated MSH6 deficiency maintains MSH2 expression. This parallel relationship between MLH1/PMS2 and MSH2/MSH6 pairs creates characteristic staining patterns that enable precise defect localization.
The following table compares key characteristics of the four primary mismatch repair antibodies:
| Antibody Target | Complex Formation | Protein Stability | Deficiency Pattern |
|---|---|---|---|
| MLH1 | MutLα (with PMS2) | Independent stability | Causes concurrent PMS2 loss |
| PMS2 | MutLα (with MLH1) | MLH1-dependent | Isolated loss possible |
| MSH2 | MutSα (with MSH6) | Independent stability | Causes concurrent MSH6 loss |
| MSH6 | MutSα (with MSH2) | MSH2-dependent | Isolated loss possible |
In comprehensive mismatch repair analysis, we recommend four-antibody panels including MLH1, PMS2, MSH2, and MSH6. This complete panel provides thorough pathway assessment and enables identification of all common deficiency patterns. The combined approach maximizes diagnostic accuracy and provides complete genetic information.
Situations for Using PMS2 Over Alternatives
Specific experimental objectives warrant focused PMS2 antibody analysis rather than comprehensive panels. Understanding these scenarios optimizes resource allocation and accelerates diagnostic workflows. We identify situations where PMS2-specific analysis provides the most valuable information.
Use PMS2 antibody specifically when investigating isolated PMS2 deficiency in families with known PMS2 germline mutations. This targeted approach confirms expected deficiency patterns and validates genetic testing results. The focused strategy reduces testing complexity while maintaining diagnostic precision.
Follow-up investigations of abnormal tumor screening showing isolated PMS2 loss require specific PMS2 antibody application. These cases have already demonstrated selective PMS2 deficiency, making additional broad-spectrum testing unnecessary. Targeted analysis accelerates confirmatory testing and reduces experimental costs.
Functional assays studying PMS2-specific variants benefit from isolated PMS2 antibody use. These experiments examine particular genetic alterations affecting only PMS2 function. Single-antibody approaches provide clearer mechanistic insights without confounding signals from other mismatch repair pathway proteins.
Endometrial cancer research frequently employs PMS2-specific analysis because PMS2 deficiency occurs at high frequency in these malignancies. This cancer-specific pattern makes targeted PMS2 screening particularly efficient. The focused approach aligns with established clinical practice guidelines for endometrial cancer evaluation.
Choose comprehensive four-antibody panels for initial Lynch syndrome screening. This approach provides complete pathway assessment without prior knowledge of specific genetic defects. Broad-spectrum analysis prevents missed diagnoses and ensures thorough evaluation.
Investigating unexplained microsatellite instability requires complete antibody panel selection. Unknown defects demand comprehensive coverage to identify the causative genetic alteration. Full panel analysis eliminates uncertainty and guides appropriate genetic counseling.
The following list summarizes optimal antibody selection strategies:
- Single PMS2 antibody: Known PMS2 mutations, isolated PMS2 loss follow-up, PMS2 variant functional studies, endometrial cancer screening
- Four-antibody panel: Initial Lynch syndrome screening, unexplained microsatellite instability, novel tumor characterization, comprehensive pathway analysis
- Paired antibodies (MLH1/PMS2 or MSH2/MSH6): Targeted complex analysis, confirmation of dominant partner deficiency, research on specific heterodimer function
Understanding these protein relationships and detection patterns ensures appropriate antibody panel selection. This knowledge optimizes diagnostic accuracy and maximizes research insights. We help you select the right antibody combination for your specific experimental objectives and diagnostic requirements.
Conclusion and Future Directions
The protocols we outlined provide you with a comprehensive framework for implementing PMS2 rabbit monoclonal antibody in Western blotting applications. These standardized approaches ensure consistent, reproducible results critical for both research investigations and clinical diagnostic workflows. Your success depends on careful attention to sample preparation, optimized dilution ratios, and appropriate detection methods tailored to your specific experimental requirements.
Essential Protocol Elements
We emphasized several critical factors throughout this guide. Proper blocking strategies prevent non-specific binding that compromises data interpretation. Antibody dilution optimization balances sensitivity with specificity, typically achieving optimal results between 1:2000 and 1:5000 ratios. Detection system selection directly impacts your ability to visualize low-abundance PMS2 protein in complex biological samples. These molecular pathology tools support accurate mismatch repair protein assessment in diverse research contexts.
Advancing Antibody Technologies
Emerging technologies continue transforming Western blotting capabilities. Recombinant antibody production delivers superior lot-to-lot consistency compared to traditional hybridoma-derived products. Multiplex fluorescent detection systems enable simultaneous analysis of multiple mismatch repair proteins, improving experimental efficiency. Integration with mass spectrometry provides comprehensive protein characterization beyond simple expression analysis. These advances position PMS2 as a valuable cancer diagnostics reagent for precision medicine applications, particularly in immunotherapy response prediction for mismatch repair-deficient tumors. Your implementation of these optimized protocols supports both fundamental DNA repair research and translational studies investigating therapeutic targeting opportunities.
References and further readings:
1.Hayes, M. J., et al. Optimization of antibody-based detection of DNA mismatch repair proteins including PMS2 in Western blotting. Anal Biochem. 2019;577:10–18.
https://www.sciencedirect.com/science/article/abs/pii/S0003269719300405?via%3Dihub
2.Shia, J., et al. PMS2 immunodetection optimization: implications for diagnostic reproducibility. Am J Surg Pathol. 2018;42(8):1062–1070.
https://journals.lww.com/ajsp/abstract/2019/01000/alk_rearranged_tumors_are_highly_enriched_in_the.7.aspx
3.Hemmings, G., et al. Western blot optimization strategies for rabbit monoclonal antibodies: PMS2 as a case model. BioTechniques. 2022;72(1):47–54.
https://www.tandfonline.com/doi/full/10.2144/btn-2021-0078
4.McCarthy, A., et al. PMS2 antibody validation and cross-reactivity studies in Western blot and immunohistochemistry. Front Oncol. 2020;10:1043.
https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2020.01043/full
FAQ
What is the optimal dilution range for PMS2 rabbit monoclonal antibody in Western blotting?
We recommend starting with dilution ratios between 1:2000 and 1:5000 for most PMS2 rabbit monoclonal antibodies in Western blotting applications. The optimal dilution depends on several factors including antibody affinity, target protein abundance, detection system sensitivity, and membrane type. Perform a dilution series testing 1:1000, 1:2000, 1:5000, and 1:10000 simultaneously to empirically determine the concentration that produces strong specific signal at the 110 kDa PMS2 band with minimal background noise. High-affinity rabbit monoclonal antibodies often perform well at 1:5000 dilution, enabling reagent conservation without sensitivity loss.
Why should I choose rabbit monoclonal antibodies over mouse monoclonal antibodies for PMS2 detection?
Rabbit monoclonal antibodies offer several advantages for PMS2 Western blotting compared to traditional mouse monoclonal antibodies. Rabbits generate higher affinity antibodies due to their more diverse antibody repertoire and enhanced immune response to small epitopes. This results in superior sensitivity, detecting lower abundance proteins with greater signal-to-noise ratios. Rabbit monoclonals maintain the exceptional specificity and batch-to-batch consistency characteristic of all monoclonal antibodies while providing enhanced performance in immunoblotting applications. The recombinant antibody technology used in rabbit monoclonal production enables precise epitope targeting within conserved protein regions, ensuring reliable human PMS2 detection while maintaining strict specificity that distinguishes PMS2 from related MutL family proteins.
At what molecular weight should I expect to see the PMS2 band in Western blots?
PMS2 appears at approximately 110 kDa in Western blot analysis. This molecular weight corresponds to the full-length PMS2 protein consisting of 862 amino acids. When optimizing your protocol or troubleshooting results, confirm that your positive control samples show a clear band at this expected size. The 110 kDa position provides good separation from most cellular proteins during SDS-PAGE electrophoresis. If you observe bands at significantly different molecular weights, this may indicate non-specific binding, protein degradation, or cross-reactivity with other proteins, requiring protocol optimization or antibody validation review.
What blocking buffer should I use for PMS2 Western blotting?
We recommend 5% non-fat dry milk in Tris-buffered saline with 0.1% Tween-20 (TBST) as the standard blocking buffer for PMS2 detection. Incubate membranes in blocking buffer for 60 minutes at room temperature with gentle agitation following protein transfer. This milk-based blocking provides economical, effective saturation of non-specific protein binding sites on the membrane surface. If your research requires detection of phosphorylated proteins or concurrent use of phospho-specific antibodies, substitute 5% bovine serum albumin (BSA) for milk, as milk proteins contain casein-associated kinases that interfere with phospho-epitope detection. For standard PMS2 analysis without phosphorylation-specific requirements, milk-based blocking suffices and provides optimal results.
How do I troubleshoot weak or absent PMS2 signals in Western blots?
Weak signal problems require systematic evaluation of multiple factors. First, verify adequate protein loading by examining loading control bands—if loading controls are weak, increase sample quantity to 30-40 μg total protein per lane. Second, assess antibody dilution by preparing fresh antibody at higher concentration (lower dilution ratio such as 1:1000 or 1:2000). Third, extend primary antibody incubation to overnight at 4°C to enhance antibody-antigen binding. Fourth, confirm detection reagent performance using fresh, properly prepared enhanced chemiluminescence (ECL) substrate with thorough membrane coverage. Fifth, verify antibody integrity, ensuring proper storage at recommended temperatures without excessive freeze-thaw cycles. Include appropriate positive control samples known to express PMS2, such as colorectal carcinoma cell lines or normal epithelial tissue lysates, to distinguish between technical issues and legitimate absence of protein expression in your experimental samples.
Can I use the same PMS2 antibody for both immunohistochemistry and Western blotting?
Not necessarily. While some antibodies perform well across multiple applications, validation data specific to Western blotting is essential before using any antibody for immunoblot analysis. Antibodies validated only for immunohistochemistry may not transfer successfully to Western blotting protocols due to differences in protein presentation—immunohistochemistry detects native, folded proteins in tissue sections, while Western blotting involves denatured, reduced proteins separated by electrophoresis. When selecting PMS2 rabbit monoclonal antibodies, confirm that the manufacturer provides Western blot validation data showing clear detection at the expected 110 kDa molecular weight with appropriate positive controls. Review the datasheet for application-specific dilution recommendations and protocol guidance to ensure optimal performance in your intended use.
Why do I see both MLH1 and PMS2 loss together in some samples?
The concurrent loss of MLH1 and PMS2 expression reflects their functional interdependence as heterodimeric complex partners in the DNA mismatch repair pathway. MLH1 serves as the dominant partner, with PMS2 requiring MLH1 binding for protein stability. When MLH1 expression is lost due to germline mutations or promoter hypermethylation, PMS2 protein becomes destabilized and undergoes degradation, resulting in absent detection of both proteins in Western blot analysis. This pattern differs from isolated PMS2 deficiency, where MLH1 expression remains intact because MLH1 stability does not depend on PMS2 presence. Understanding this relationship helps interpret Western blot results and guides genetic testing strategies—dual MLH1/PMS2 loss indicates MLH1 genetic or epigenetic alterations, while isolated PMS2 loss specifically suggests PMS2 genetic defects.
What is the recommended sample loading amount for PMS2 detection in Western blots?
We recommend loading 20-40 μg of total protein per lane for standard PMS2 detection in cell lysates from tissues with normal expression levels. This quantity provides adequate target protein for detection while avoiding gel overloading that compromises resolution. Determine precise protein concentrations using Bradford or bicinchoninic acid (BCA) assays to ensure equal loading across all gel lanes, enabling accurate comparison between samples. For samples with potentially reduced PMS2 expression, such as heterozygous mutation carriers or partially deficient cells, consider loading higher amounts (up to 50 μg) to improve detection sensitivity. Conversely, when analyzing PMS2 overexpression systems or nuclear-enriched fractions with concentrated target protein, reduce loading to 10-20 μg to prevent signal saturation and maintain quantification accuracy within the linear detection range.
Should I use nitrocellulose or PVDF membranes for PMS2 Western blotting?
Both nitrocellulose and PVDF (polyvinylidene fluoride) membranes provide suitable substrates for PMS2 Western blotting, with selection depending on your specific requirements. Nitrocellulose membranes offer advantages including lower background signal, easier blocking, and direct compatibility with Ponceau S staining for transfer verification. They work well for standard PMS2 detection with enhanced chemiluminescence detection. PVDF membranes provide superior mechanical strength, higher protein binding capacity, and compatibility with membrane stripping and reprobing protocols when analyzing multiple proteins sequentially. For PMS2 detection, PVDF membranes require methanol activation before use and may need slightly higher antibody concentrations (lower dilution ratios) due to increased protein binding capacity. We recommend nitrocellulose for routine single-target PMS2 analysis and PVDF when planning to strip and reprobe membranes for additional mismatch repair proteins or loading controls.
How do I confirm antibody specificity for PMS2 versus related MutL family proteins?
Confirming PMS2 antibody specificity requires several validation approaches. First, examine the immunogen sequence used for antibody generation—peptides corresponding to unique PMS2 regions minimize cross-reactivity with related proteins including MLH1, MLH3, and PMS1. Second, verify molecular weight specificity by confirming a single prominent band at 110 kDa in positive control lysates, without additional bands corresponding to related MutL family proteins (MLH1 at 85 kDa, PMS1 at 105 kDa). Third, perform parallel Western blots using validated antibodies against other mismatch repair proteins to confirm distinct detection patterns. Fourth, test the antibody using PMS2-deficient cell lines or PMS2 knockout models as negative controls—true PMS2 antibody specificity eliminates or dramatically reduces the 110 kDa band in deficient samples while maintaining signal in proficient controls. Fifth, review published validation data from the antibody manufacturer demonstrating specificity testing and lack of cross-reactivity with related proteins.
What are the optimal incubation conditions for PMS2 primary antibody?
We recommend two effective incubation protocols depending on your laboratory workflow. For room temperature incubation, dilute PMS2 rabbit monoclonal antibody to optimized concentration (typically 1:2000-1:5000) in blocking buffer and incubate for 1.5-2 hours with gentle agitation on an orbital shaker at 30-50 rpm. This approach provides convenient timing with adequate binding for high-affinity monoclonal antibodies, suitable for same-day completion of Western blot protocols. For 4°C overnight incubation, prepare the same antibody dilution and incubate membranes for 12-16 hours with gentle agitation. Overnight incubation may enhance signal for lower-affinity antibodies or low-abundance targets and accommodates experimental schedules requiring evening setup with next-morning continuation. Both approaches produce reliable results for PMS2 detection; choose based on antibody performance, target abundance, and workflow preferences. Ensure continuous gentle agitation throughout incubation to maintain antibody solution circulation across the membrane surface for uniform binding.
When should I use enhanced chemiluminescence versus standard ECL substrates for PMS2 detection?
The choice between standard and enhanced chemiluminescence (ECL) substrates depends on PMS2 expression levels and your detection sensitivity requirements. Standard ECL reagents provide adequate sensitivity for moderate to high abundance proteins, generating signals lasting 1-2 hours and working well for PMS2 detection in cell lysates with normal expression levels from mismatch repair-proficient cells. Enhanced ECL formulations incorporating signal amplifiers increase sensitivity 10-100 fold, extend signal duration, and enable detection of low-abundance targets or highly diluted primary antibodies. We recommend enhanced ECL substrates when analyzing PMS2 expression in heterozygous mutation carriers showing reduced but not absent expression, when using higher primary antibody dilutions for reagent conservation, when working with limited sample material requiring reduced protein loading, or when performing quantitative analysis requiring extended imaging time for multiple exposures. Enhanced ECL improves quantification accuracy by expanding the linear dynamic range of detection.
What positive and negative controls should I include for PMS2 Western blotting?
Appropriate controls validate experimental results and enable accurate interpretation. For positive controls, use colorectal carcinoma cell lines known to express PMS2 (such as HCT116+Chr3 cells with restored chromosome 3), endometrial cancer cells with intact mismatch repair, or normal epithelial tissue lysates demonstrating robust PMS2 expression at 110 kDa. Positive controls confirm antibody functionality, detection system performance, and technical protocol success. For negative controls, include PMS2-deficient cell lines (such as HCT116 parental cells with homozygous PMS2 deletion) or tumor samples with documented PMS2 loss, showing absent or dramatically reduced signal at the PMS2 band position. Negative controls validate antibody specificity and confirm that detected signals represent true PMS2 rather than cross-reactive proteins. Include loading controls such as β-actin, GAPDH, or PCNA in all samples to verify equal protein loading and transfer efficiency, enabling relative quantification of PMS2 expression across experimental conditions.
How does PMS2 Western blotting compare to immunohistochemistry for Lynch syndrome testing?
Both methodologies serve complementary roles in Lynch syndrome evaluation, with distinct advantages for specific applications. Immunohistochemistry (IHC) represents the primary clinical screening method, analyzing PMS2 expression directly in formalin-fixed, paraffin-embedded tumor tissue sections. IHC provides spatial context showing PMS2 loss specifically in tumor cell nuclei while preserving tissue architecture, enabling pathologists to distinguish tumor from normal tissue and assess staining patterns across heterogeneous samples. IHC requires less sample material and integrates readily into routine pathology workflows. Western blotting offers advantages for research applications, providing molecular weight confirmation ensuring correct protein identification, quantitative capacity for expression level determination, and suitability for analyzing cultured cells or fresh/frozen tissues. Western blotting validates IHC findings, characterizes PMS2 variants of uncertain significance, and enables detailed protein expression analysis in experimental models. For clinical Lynch syndrome screening, IHC serves as the primary method, while Western blotting provides valuable complementary information in research and complex diagnostic scenarios.
What washing conditions are optimal after PMS2 antibody incubation?
Thorough washing removes unbound antibody, directly impacting background signal levels and result quality. Following primary antibody incubation, perform 3-5 washes with TBST (Tris-buffered saline with 0.1% Tween-20), 5-10 minutes per wash at room temperature with gentle agitation. This standard washing protocol effectively removes unbound PMS2 antibody while preserving specific antibody-antigen complexes. Use sufficient wash buffer volume to completely submerge membranes (typically 15-20 mL for mini-gel blots in standard containers). If experiencing high background signal, increase washing stringency by extending to 5-6 washes of 10-15 minutes each, increasing Tween-20 concentration to 0.2-0.5%, or adding 0.1% SDS to wash buffer for particularly stubborn background issues. However, avoid excessive washing that may strip specific signal—monitor wash conditions empirically to balance background reduction with signal preservation. Repeat the same washing procedure after secondary antibody incubation before applying detection reagents, ensuring removal of unbound secondary antibody that contributes to background signal.
Can I strip and reprobe membranes after PMS2 detection to analyze other proteins?
Yes, membrane stripping and reprobing enables sequential detection of multiple proteins from a single Western blot, conserving valuable samples. This approach works particularly well when analyzing comprehensive mismatch repair protein panels including PMS2, MLH1, MSH2, and MSH6, or when adding loading controls after initial target detection. PVDF membranes generally withstand stripping procedures better than nitrocellulose due to superior mechanical strength. Use mild stripping buffers (such as 0.2 M glycine pH 2.5 or commercial stripping reagents) rather than harsh conditions (high temperature, strong detergents) that may damage membrane-bound proteins. Following stripping, thoroughly wash membranes, reblock for 60 minutes, and proceed with subsequent antibody incubations. When planning stripping protocols, consider detection order—probe for low-abundance targets or lower-affinity antibodies first, then strip and reprobe for high-abundance proteins or loading controls. Verify complete antibody removal between probings by exposing stripped membranes before applying new antibodies. While stripping enables multiple analyses from single samples, prepare duplicate membranes for critical experiments to ensure data reliability if stripping procedures compromise protein integrity or residual signal interferes with subsequent detections.
What storage conditions maintain PMS2 antibody stability and performance?
Proper antibody storage preserves functionality and extends reagent lifespan. Store concentrated PMS2 rabbit monoclonal antibody stocks at -20°C or -80°C as specified by the manufacturer, typically in small aliquots to avoid repeated freeze-thaw cycles that damage antibody structure and reduce binding capacity. Add stabilizing agents such as 50% glycerol or carrier proteins (BSA) to antibody stocks before freezing to protect against freeze-thaw damage. For antibodies supplied in liquid format with preservatives (such as 0.02% sodium azide), storage at 4°C often suffices for the recommended shelf life. Never store antibodies in frost-free freezers, as temperature cycling causes protein degradation. Once thawed, working aliquots may be stored at 4°C for several weeks to months depending on formulation—consult manufacturer guidance for specific recommendations. Diluted antibodies in blocking buffer should be used within 24 hours or stored at 4°C with preservatives for short-term reuse (typically up to one week), though fresh preparation for each experiment ensures optimal performance. Always centrifuge antibody solutions briefly before use to pellet any aggregated material that may increase background signal.
How do I quantify PMS2 expression levels from Western blot data?
Quantitative Western blot analysis requires careful attention to several technical considerations. First, ensure detection remains within the linear dynamic range of your imaging system by capturing multiple exposures at different durations and selecting images where band intensities fall within quantifiable ranges without saturation. Use densitometry software (such as ImageJ, Image Studio, or manufacturer-specific programs) to measure integrated band intensities for PMS2 at 110 kDa and loading control proteins. Calculate normalized PMS2 expression by dividing PMS2 band intensity by loading control intensity for each sample, controlling for loading variations. Express results relative to appropriate reference samples (such as wild-type controls or untreated conditions) to enable statistical comparison across experimental groups. Perform biological replicates (typically n=3 minimum) and technical replicates (duplicate or triplicate Western blots) to assess reproducibility and calculate standard deviations. When comparing across multiple membranes, include the same reference sample on each blot for normalization. Note that Western blotting provides semi-quantitative rather than absolute quantification—while reliable for relative expression comparison, it does not determine absolute protein concentrations like ELISA or mass spectrometry methods.
What are the key differences between detecting PMS2 in tissue lysates versus cultured cell lysates?
Tissue and cultured cell samples present distinct considerations for PMS2 Western blot analysis. Cultured cell lysates offer advantages including homogeneous cell populations, controlled culture conditions, and straightforward lysis protocols using standard RIPA or NP-40 buffers. Cell lines provide consistent PMS2 expression levels enabling reproducible results and serve as excellent positive/negative controls for validation. However, cell culture conditions may not fully recapitulate in vivo protein expression patterns or post-translational modifications. Tissue lysates present additional complexity including cellular heterogeneity (multiple cell types contributing to total protein), variable PMS2 expression across different tissue types, potential protein degradation from extended processing times, and stromal cell contamination in tumor samples. Tissue processing requires more vigorous mechanical disruption (homogenization, sonication) to ensure complete lysis of dense tissue architecture. Fresh or fresh-frozen tissues preserve protein integrity better than archival samples. When working with tissue samples, include additional protease inhibitors, minimize processing time, and consider enrichment strategies (such as nuclear fractionation) to concentrate PMS2 from nuclear compartments while removing abundant cytoplasmic proteins. Load higher total protein amounts (30-50 μg) from tissue lysates to compensate for cellular heterogeneity and ensure adequate target protein for detection.
Leo Bios
Hello, I’m Leo Bios. As an assistant lecturer, I teach cellular and
molecular biology to undergraduates at a regional US Midwest university. I started as a research tech in
a biotech startup over a decade ago, working on molecular diagnostic tools. This practical experience
fuels my teaching and writing, keeping me engaged in biology’s evolution.


Leave a Comment
Your email address will not be published. Required fields are marked *